
Dr. Kala Bharath Pilla
I am currently working as Senior Computational Biologist at T-Therapeutics headed by Prof. Allan Bradley in Cambridge, U.K.
My previous roles include a Postdoctoral fellowship in integrative structural modelling under the mentorship of Prof. Andrej Sali at University of California in San Francisco and
a doctoral fellowship in computational structural biology under the supervision of Prof. Thomas Huber and Prof. Gottfried Otting at the Australian National University in Canberra
My thesis was on “Protein Structure Determination by Paramagnetic NMR and Computational Hybrid Approach”.
My research interests are in developing new algorithms for integrative/hybrid modeling of proteins and protein complexes, protein structure prediction and protein engineering/design.
My notable contributions:
“DINGO-PCS/RDC-NOE” : 3D assembly of Individual smotifs to Near-native Geometry as Orchestrated by PCSs (or) RDCs and NOEs
“GPS-Rosetta” : Protein Structure determination inspired by GPS algorithms.
Research
“DINGO-PCS” : 3D assembly of Individual smotifs to Near-native Geometry as Orchestrated by PCSs
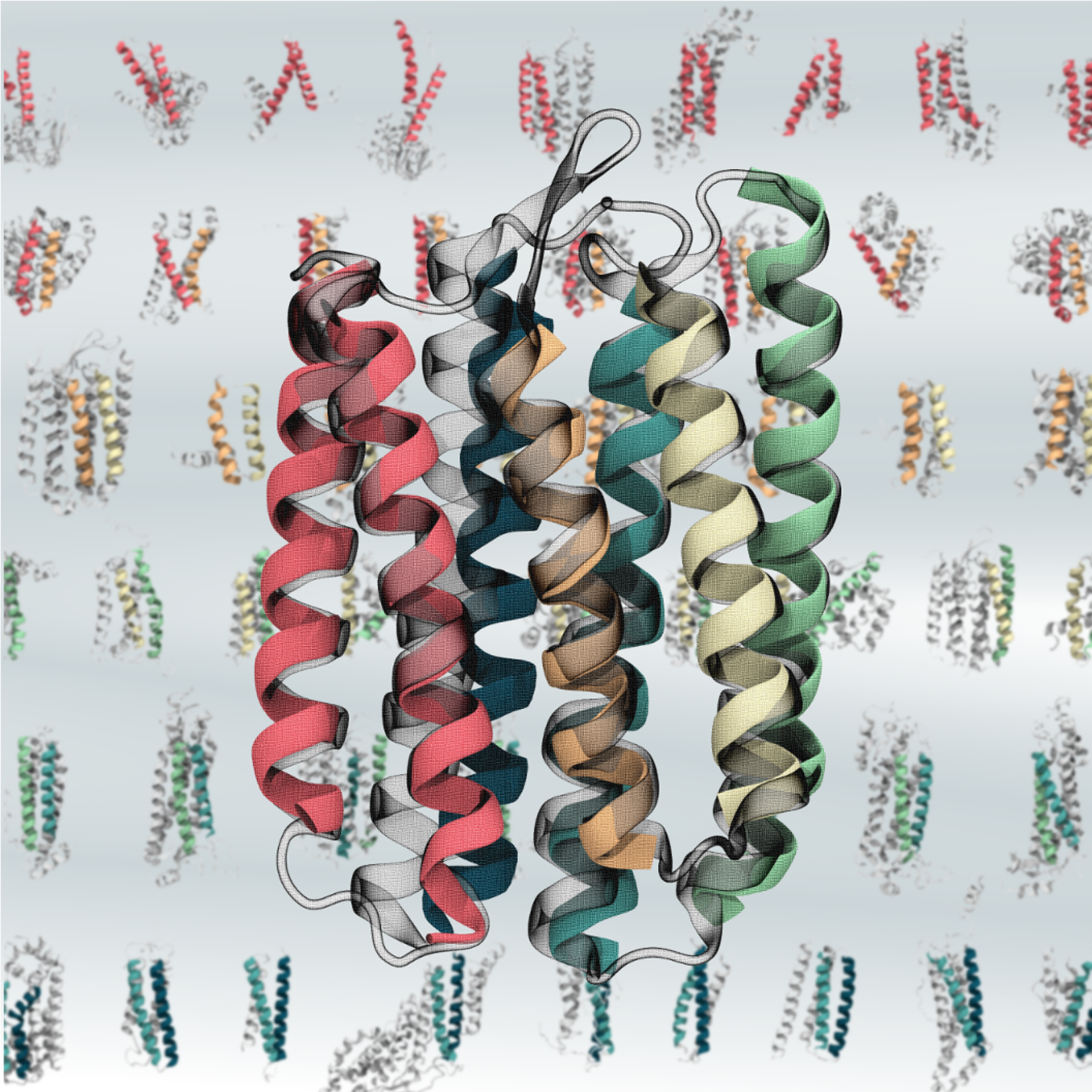
Computational and NMR hybrid approaches have proven to be a rapid and reliable approach for 3D structure determination of small proteins, but the current state-of-the-art algorithms struggle to perform with larger proteins. In this work, we demonstrate a new computational algorithm that assembles the 3D structure of a protein from its constituent super-secondary structural motifs (Smotif) with the help of pseudocontact (PCS) restraints for backbone amide protons, where the PCSs are produced from different metal centers. The algorithm, DINGO-PCS (3D assembly of Individual Smotifs to Near-native Geometry as Orchestrated by PCSs), employs the PCSs to recognize, orient and assemble the constituent Smotifs of the target protein without using any other experimental measurements or computational force fields. Using a universal Smotif database that applies to any target protein, the DINGO-PCS algorithm is designed to exhaustively enumerate any given Smotif. We benchmarked the DINGO-PCS algorithm against a set of ten different protein targets ranging from 100-220 residues with different topologies. For nine out of these ten targets, the method was able to identify near native Smotifs.
Protein Structure Determination by Assembling Super-Secondary Structure Motifs Using Pseudocontact Shifts
KB Pilla, G Otting, T Huber.
Structure 25 (3), 559-568, 2017.
Github Download:https://github.com/kalabharath/DINGO-PCS
Identifying Binding Mode of Ligands by Computaional and Paramagnetic-NMR Hybrid Approach
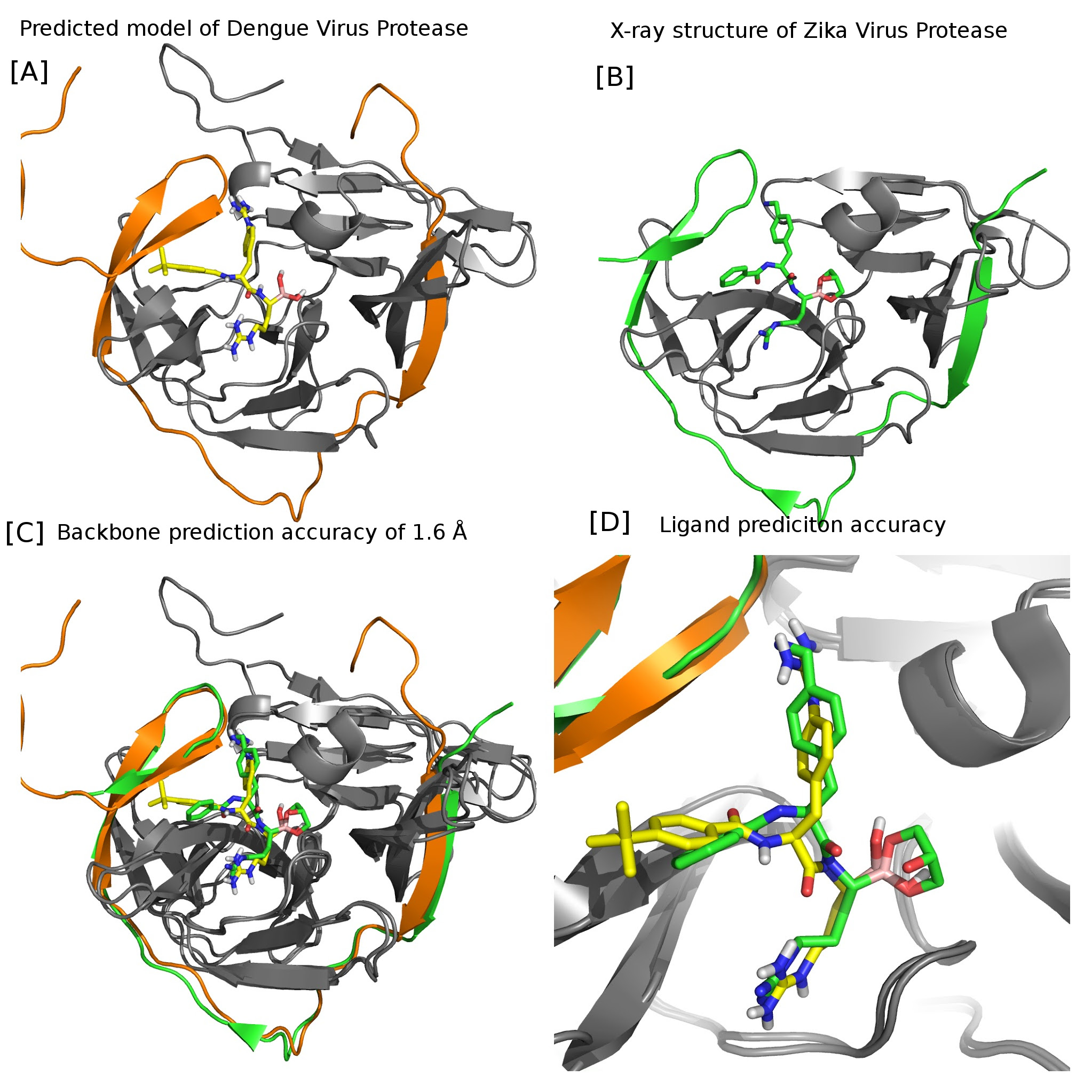
(A) Computational model of Ligand (peptidomimetic broronate inhibitor) bound to Dengue virus protease, established using PCSs. (Published on March 2016). (B) Crystal structure of Similar (peptidomimetic broronate inhibitor) bound to Zika Virus protease. PDBID:5LC0 (published on July 2016). (C) C-alpha RMSD between computational model Vs X-ray structure is 1.66 Angstrom. Note: Computational model was blind and published ahead of X-ray structure. (D) Close-up view of orientation of the peptidomimetic broronate inhibitors. Computational model generated using experimental PCSs (yellow). X-ray model (green)
Structure-guided drug design relies on detailed structural knowledge of protein–ligand complexes, but crystallization of cocomplexes is not always possible. Here we present a sensitive nuclear magnetic resonance (NMR) approach to determine the binding mode of tightly binding lead compounds in complex with difficult target proteins. In contrast to established NMR methods, it does not depend on rapid exchange between bound and free ligand or on stable isotope labeling, relying instead on a tert-butyl group as a chemical label. tert-Butyl groups are found in numerous protein ligands and deliver an exceptionally narrow and tall 1H NMR signal. We show that a tert-butyl group also produces outstandingly intense intra- and intermolecular NOESY cross-peaks. These enable measurements of pseudocontact shifts generated by lanthanide tags attached to the protein, which in turn allows positioning of the ligand on the protein. Once the ligand has been located, assignments of intermolecular NOEs become possible even without prior resonance assignments of protein side chains. The approach is demonstrated with the dengue virus NS2B-NS3 protease in complex with a high-affinity ligand containing a tert-butyl group.
Sensitive NMR approach for determining the binding mode of tightly binding ligand molecules to protein targets
WN Chen, C Nitsche, KB Pilla, B Graham, T Huber, CD Klein, G Otting
Journal of the American Chemical Society(2016), 138 (13), 4539-4546
Iterative Re-sampling Technique for 3D Structure Determinations of Large Proteins
Incorporating sparse NMR restraints in structure prediction methods, such as Rosetta, can guide the sampling towards the global energy minimum. However, the approach is limiting with the increase in the size of the protein. Here, we demonstrate a new computational algorithm which improves Rosetta’s fragment assembly method that utilizes long-range structural information encoded in Pseudocontact shifts (PCS) generated from site-specific attachment of paramagnetic lanthanide ions at four different sites. The algorithm relies solely on the sparse PCS datasets and utilizes them in multiple different means to determine the 3D structure of a protein. Firstly, the algorithm directs the conformational search of the proteins towards the native state; secondly, it ranks best structural models from fitted PCS restraints; thirdly, it identifies native like local structures and rebuilds the fragment libraries. Finally, the algorithm iterates the sampling using the PCS derived fragment libraries until convergence. We show that our PCS driven iterative resampling algorithm is quite powerful in obtaining accurate models over a benchmark set of eight different proteins, ranging from 100 to 220 residues, using only backbone amide hydrogen PCSs. The iterative resampling algorithm is strictly data dependent and data driven and poses a powerful alternative to not only traditional approaches but also the other existing resampling based methods for 3D structure determination.
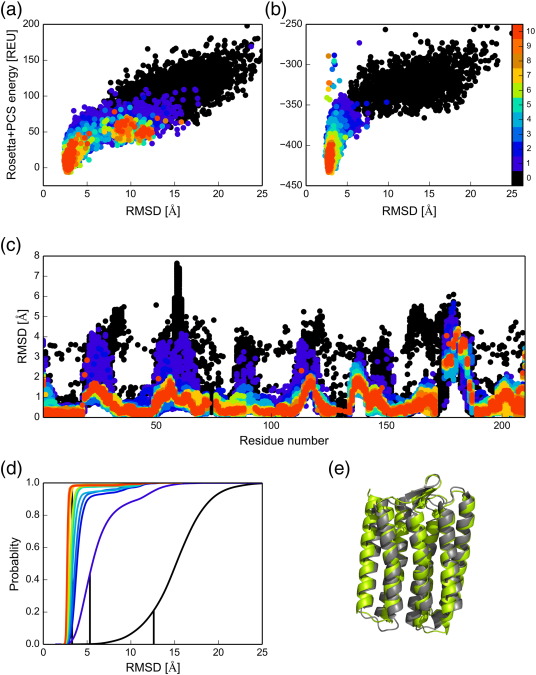
Results of the iterative GPS-Rosetta on a 200 residue trans-membrane bacterial rhodopsin protein, pSRII,PDBID:1H68. Structures showing the combined Rosetta centroid energy and PCS energy in (A) and Rosetta all-atom energy and PCS energy in (B) versus the Cα RMSD. (C) Improvement in quality of fragments identified by overlapping Δχ-tensors in PCS driven iterative scheme. For each nine-residue fragment, the RMSD is calculated between the fragment and its corresponding native fragment in the crystal structure. The quality of the standard fragment library used for zeroth iteration is shown in black and the PCS identified fragments in successive iterations are color-coded from blue to red.
Pseudocontact shift-driven iterative resampling for 3D structure determinations of large proteins.
KB Pilla, G Otting, T Huber.
J. Mol. Biol. 428, 522-532 (2016) pdf Supp pdf
Capturing Conformational States
Understanding induced conformational changes at an atomic level is critical for drug development. We identify these conformational states using sparse experimental data from paramagnetic NMR spectroscopy. We demonstrate this using our GPS-Rosetta approach on the example of the Dengue virus protease structure which can exist in two forms, a “closed” form in the presence of an inhibitor and an “open” form without inhibitor. Our approach succeeds in the structural elucidation of both forms while the traditional computational method fails.
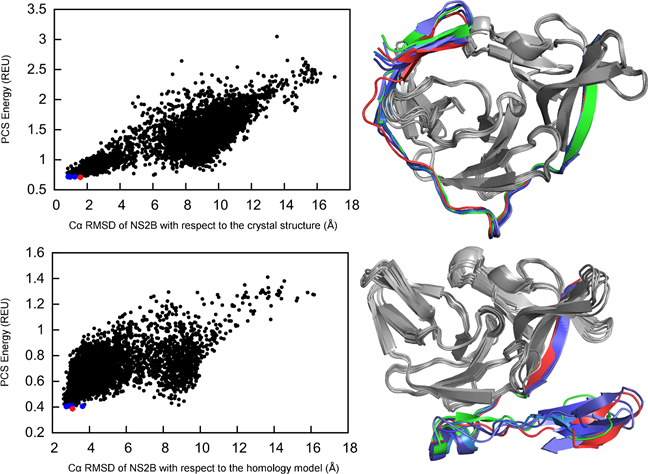
Capturing Conformational States in Proteins Using Sparse Paramagnetic NMR Data.
KB Pilla, J Koehler Leman, G Otting, T Huber.
PLoS ONE 10(5): e0127053 2015 link to open access article
GPS-Rosetta
Paramagnetic ions attached to proteins via chemical chelating tags can act as GPS satellites. PCS of a nuclear spin from a paramagnetic metal ion ties its location to a certain isosurface, a second PCS measured for the same nucleus from a paramagnetic ion attached at a different site restricts the spin to lie on intersecting isosurfaces. A third PCS measured from a paramagnetic ion attached at a site different from the first two would further restrict the location of the spin in space. This technique is analogous to the method of finding location on Earth from three or more GPS satellites. This method is incorporated into the Rosetta fragment assembly framework and dramatically alters the conformational sampling, directing the search towards the native. The GPS-Rosetta algorithm works with PCSs recorded in either solution state or solid state.
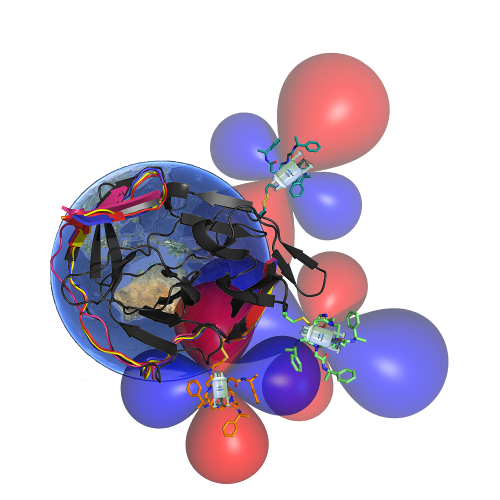
Three-dimensional protein fold determination from backbone amide pseudocontact shifts generated by lanthanide tags at multiple site.
^H Yagi, ^KB Pilla, A Maleckis, B Graham, T Huber, G Otting. (^ co first)
Structure 21 (6), 883-890 11 2013. pdf
Magic angle spinning NMR structure determination of proteins from pseudocontact shifts.
J Li, KB Pilla, Q Li, Z Zhang, X Su, T Huber, J Yang.
Journal of the American Chemical Society 135 (22), 8294-8303, 11 2013. pdf
Publications
Research Articles
Link to my Google Scholar profile.
17. Recognizing amino acid sidechains in a medium resolution cryo-electron density map
D. Mondal, V. Kumar, T. Satler, R. Ramachandran, D. Saltzberg, I. Chemmama, K.B. Pilla, I. Echeverria, B.M. Webb, M. Gupta, K.A. Verba, A. Sali.
bioRxiv, 2024.
16. Soluble TREM2 inhibits secondary nucleation of Aβ fibrillization and enhances cellular uptake of fibrillar Aβ
K.D. Belsare, H. Wu, D. Mondal, A. Bond, E. Castillo, J. Jin, H. Jo, A.E. Roush, K.B. Pilla, A. Sali, C. Condello, W.F. DeGrado.
Proc Natl Acad Sci USA 119, e2114486119, 2022.
15. Bayesian metamodeling of complex biological systems across varying representations
B. Raveh, L. Sun, K.L. White, T. Sanyal, J. Tempkin, D. Zheng, K.B. Pilla, J. Singla, C. Wang, J. Zha, A. Li, N.A. Graham, C. Kesselman, R.C. Stevens, A. Sali
Proc Natl Acad Sci USA 118, e2104559118, 2021,
14. Macromolecular modeling and design in Rosetta: recent methods and frameworks
J.K Leman, B.D Weitzner,[..] K.B. Pilla et al.
Nature methods 17 (7), 665-680
13. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing
D.E. Gordon, G.M. Jang.[..] K.B. Pilla et al.
Nature 583 (7816), 459-468
12. SSEThread: Integrative threading of the DNA-PKcs sequence based on data from chemical cross-linking and hydrogen deuterium exchange
D.J. Saltzberg, M. Hepburn, K.B Pilla, DC Schriemer, SP Lees-Miller, T.L. Blundell, A. Sali
Progress in biophysics and molecular biology 147, 92-102
11. Protein Fold Determination by Assembling Extended Super-Secondary Structure Motifs Using Limited NMR Data
K.B. Pilla
bioRxiv, 509356
10. Molecular modeling of biomolecules by paramagnetic NMR and computational hybrid methods
K.B. Pilla, K. Gaalswyk, J.L. MacCallum
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 1865, 1654-1663 (2017)
9. Protein structure determination by assembliing super-secondary structure motifs using pseudocontact shifts
K. B. Pilla, G. Otting, T. Huber
Structure 25, 559-568 (2017)
8. Sensitive NMR approach for determining the binding mode of tightly binding ligand molecules to protein targets
W.-N. Chen, C. Nitsche, K. B. Pilla, B. Graham, T. Huber, C. D. Klein, G. Otting
J. Am. Chem. Soc, 138, 4581-4586 (2016)
7. Pseudocontact shift-driven iterative resampling for 3D structure determinations of large proteins
K. B Pilla, G. Otting, T. Huber.
J. Mol. Biol. 428, 522-532 (2016) pdf Supp pdf
6. 3D Computational Modelling of Proteins Using Sparse Paramagnetic NMR Data
K.B. Pilla, G. Otting, T. huber.
Bioinformatics: Volume II: Structure, Function, and Applications, 3-21
5. Capturing Conformational States in Proteins Using Sparse Paramagnetic NMR Data
K.B. Pilla, J. Koehler Leman, G. Otting, T. Huber.
PLoS ONE 10(5): e0127053 2015 link to open access article
4.Three-dimensional protein fold determination from backbone amide pseudocontact shifts generated by lanthanide tags at multiple site
^H. Yagi, ^K.B. Pilla, A. Maleckis, B. Graham, T. Huber, G. Otting. (^ co first)
Structure 21 (6), 883-890 11 2013. pdf
3.Magic angle spinning NMR structure determination of proteins from pseudocontact shifts
J. Li, K.B. Pilla, Q. Li, Z. Zhang, X. Su, T. Huber, J. Yang.
J. Am. Chem. Soc, 135 (22), 8294-8303, 11 2013. pdf
2. InPACdb Indian plant anticancer compounds database.
U. Vetrivel, N. Subramanian, K.B. Pilla.
Bioinformation 4 (2), 71 10 2009. link to open access article
1. Open discovery: An integrated live Linux platform of Bioinformatics tools
U. Vetrivel, K.B. Pilla.
Bioinformation 3 (4), 144 7 2008. link to open access article
Contact
Contact
Email: kalabharath gmail dt com
Twitter: @kalabharath